Quickstart#
Packages#
Note: To run the spatial analysis at the end of this tutorial you will need to install ParTIpy with the tutorial extras: pip install partipy[extra].
from pathlib import Path
import scanpy as sc
import pandas as pd
import plotnine as pn
import decoupler as dc
import numpy as np
from scipy.spatial.distance import cdist
import partipy as pt
Note: Importing partipy for the first time after installing might take some time (~30s) because numba needs to compile some code in the background. After that, it’ll be quick!
Data#
First, we need to load and cache a single-cell dataset. In this example, we use mouse hepatocytes profiled by scRNA-seq from [BenMosheVM+22]. We downloaded the raw data from Zenodo and saved it to figshare for convenient access in this tutorial.
adata = sc.read("data/hepatocyte_processed.h5ad", backup_url="https://figshare.com/ndownloader/files/59459459")
adata
AnnData object with n_obs × n_vars = 1999 × 8354
obs: 'cell_type', 'zone', 'run_id', 'time_point', 'UMAP_X', 'UMAP_Y'
Preprocessing#
Before conducting archetypal analysis (AA) on single-cell RNA-seq data, it is beneficial to reduce the dimensionality of the data, as this helps remove noise and substantially increases the computational efficiency of AA.
To achieve this, we first scale the counts across all cells to a common value using sc.pp.normalize_total, followed by logarithmic normalization with sc.pp.log1p.
By default, normalize_total scales each cell to the median total counts across all cells, which preserves the typical range of expression values prior to logarithmic transformation.
Next, we identify highly variable genes and apply principal component analysis (PCA) using only these genes.
All preprocessing steps are performed using functions from the scanpy package.
sc.pp.normalize_total(adata)
sc.pp.log1p(adata)
sc.pp.highly_variable_genes(adata)
sc.pp.pca(adata, mask_var="highly_variable")
adata.layers["z_scaled"] = sc.pp.scale(adata.X, max_value=10, copy=True)
sc.pl.pca_scatter(adata, color=["Alb", "Cyp2e1", "Apoa2"])
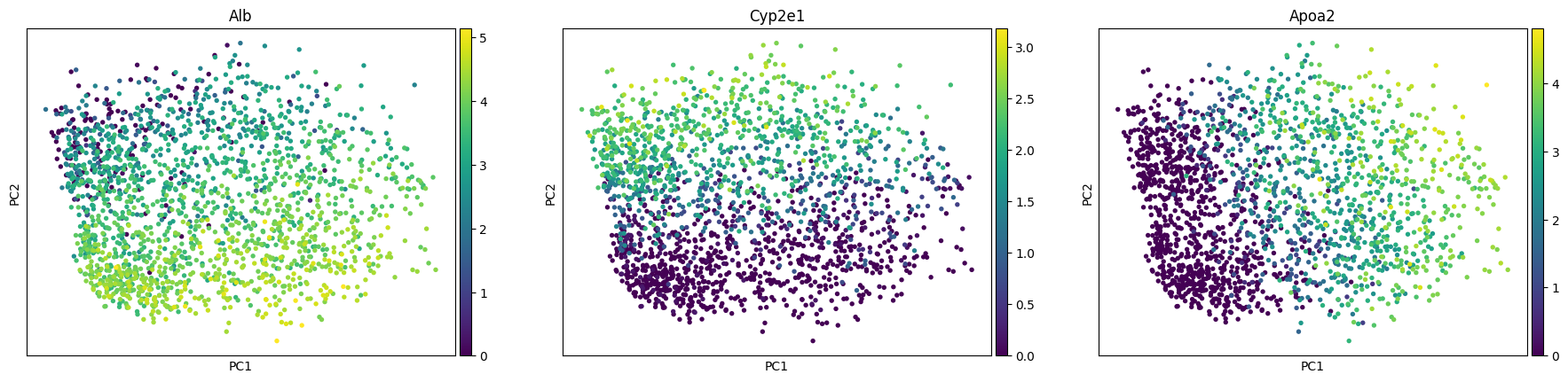
To determine the number of principal components to use for fitting the archetypes, we will examine the scree plot from the PCA. Additionally the plot shows the variance explained if we shuffle the data before computing the PCA. This plot indicates that we should only use the first three principal components.
pt.compute_shuffled_pca(adata, mask_var="highly_variable")
pt.plot_shuffled_pca(adata)
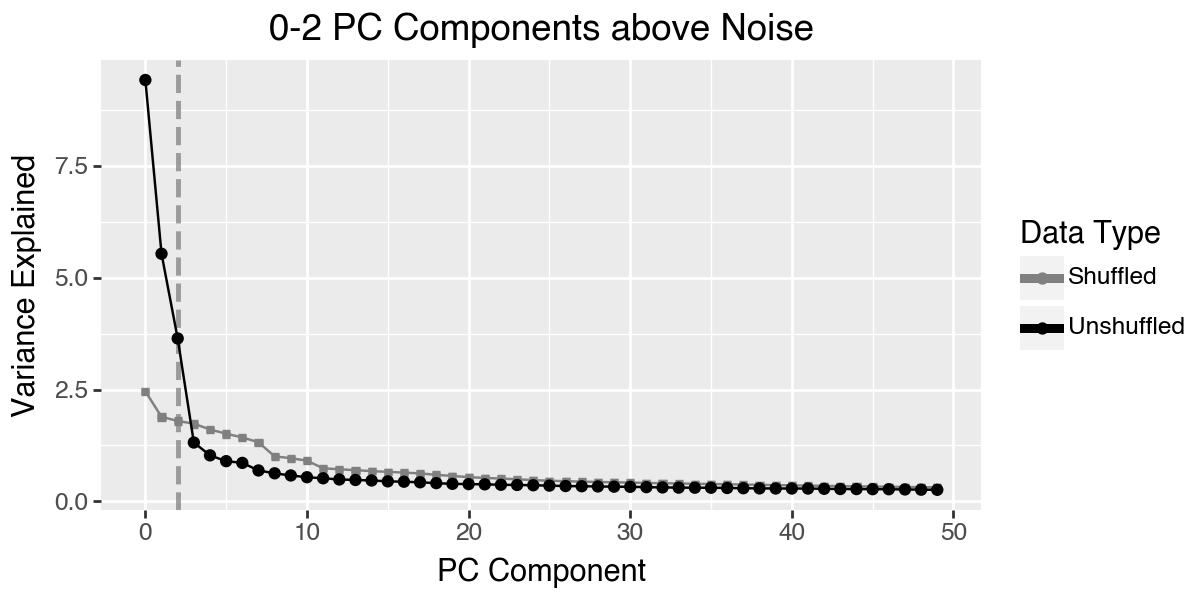
We store this setting in the adata.uns slot using the utility function set_obsm. Using n_dimensions=3 is equivalent to specifying n_dimensions=[0, 1, 2]. If you do not want to use the first principal component, for example because it captures some technical effect, then you can simply specify n_dimensions=[1, 2].
pt.set_obsm(adata=adata, obsm_key="X_pca", n_dimensions=3)
Choosing the Number of Archetypes#
Now, we are ready to perform archetypal analysis, but the key question remains: how many archetypes should be used?
There is no definitive ground truth for this choice, but several heuristics can guide the decision. A practical approach is to evaluate selection metrics across a range of archetype counts.
The function pt.compute_selection_metrics() evaluates multiple values of k (here via n_archetypes_list=range(2, 9)) and computes both variance explained and an information criterion suggested by [Sul17].
The results are cached in adata.uns["AA_selection_metrics"] and can be visualized with pt.plot_var_explained(adata) and pt.plot_IC(adata). Access is provided by the getter function pt.get_aa_metrics.
In this example, both diagnostics suggest that choosing 4 archetypes is a reasonable trade-off, since additional variance explained beyond this point is small.
adata
AnnData object with n_obs × n_vars = 1999 × 8354
obs: 'cell_type', 'zone', 'run_id', 'time_point', 'UMAP_X', 'UMAP_Y'
var: 'highly_variable', 'means', 'dispersions', 'dispersions_norm'
uns: 'log1p', 'hvg', 'pca', 'AA_pca', 'AA_config'
obsm: 'X_pca'
varm: 'PCs'
layers: 'z_scaled'
pt.compute_selection_metrics(adata=adata, n_archetypes_list=range(2, 9))
pt.summarize_aa_metrics(adata)
| k | n_archetypes | n_restarts | seed | varexpl | IC | RSS | |
|---|---|---|---|---|---|---|---|
| 0 | 2 | 2 | 5 | 42 | 0.505511 | 4112.441228 | 2965.446045 |
| 1 | 3 | 3 | 5 | 42 | 0.780190 | 4045.360678 | 1318.203125 |
| 2 | 4 | 4 | 5 | 42 | 0.933783 | 4357.642608 | 397.105713 |
| 3 | 5 | 5 | 5 | 42 | 0.968807 | 5013.518826 | 187.063721 |
| 4 | 6 | 6 | 5 | 42 | 0.986954 | 5593.265824 | 78.237045 |
| 5 | 7 | 7 | 5 | 42 | 0.990262 | 6457.961982 | 58.399693 |
| 6 | 8 | 8 | 5 | 42 | 0.992343 | 7321.808964 | 45.918877 |
pt.plot_var_explained(adata)
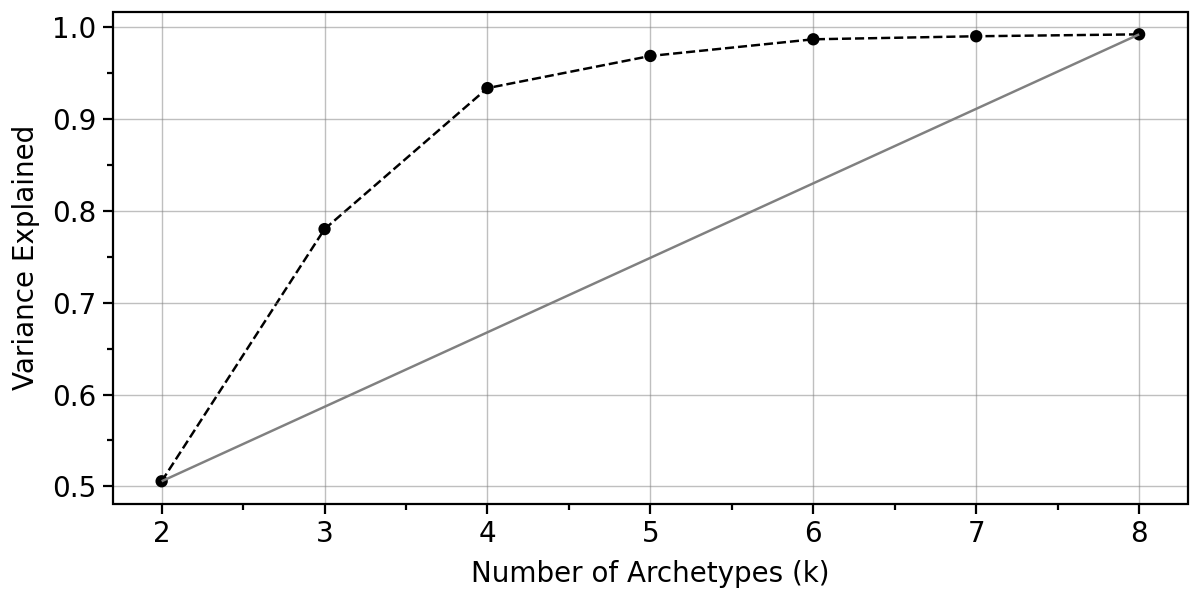
pt.plot_IC(adata)

An important consideration is whether the positions of the archetypes remain stable for a given number of archetypes. To assess this, we apply bootstrapping and perform Archetypal Analysis (AA) on resampled datasets.
By default, this function utilizes all available CPU cores, as specified by the n_jobs argument, which is set to -1. The results are stored in adata.uns["AA_bootstrap"] and can be visualized using pt.plot_bootstrap_variance(adata).
As hinted to above, this result also suggest that 4 archetypes are highly stable.
pt.compute_bootstrap_variance(adata=adata, n_bootstrap=50, n_archetypes_list=range(2, 9))
pt.plot_bootstrap_variance(adata)
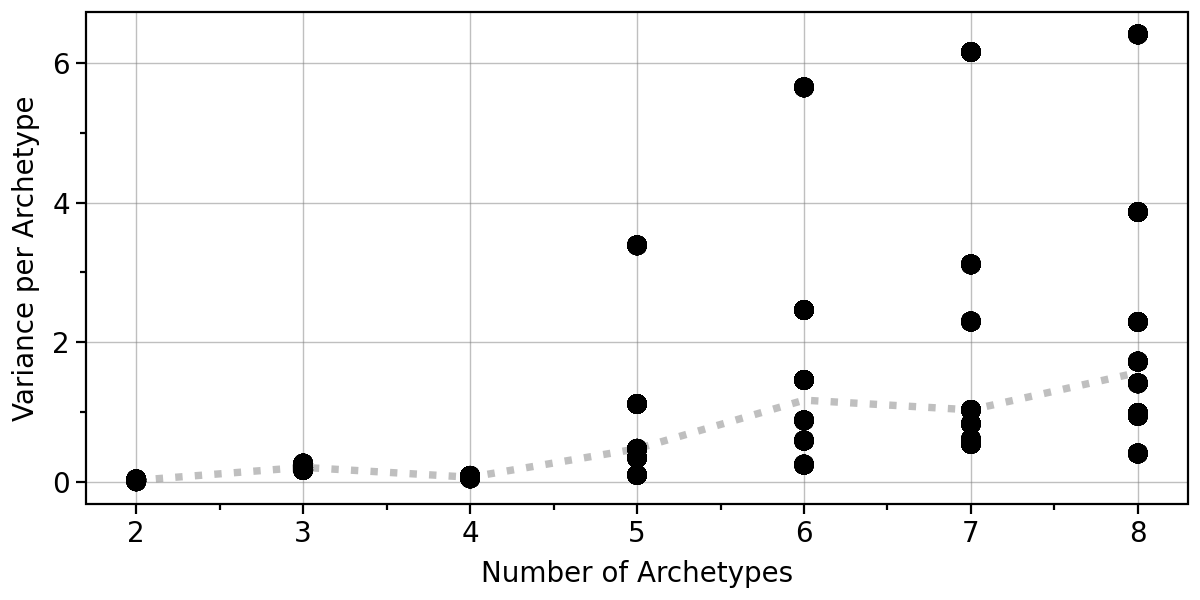
# extract the bootstrap results using pt.get_aa_bootstrap
df_list = []
for n_archs in range(2, 9):
df_list.append(pt.get_aa_bootstrap(adata, n_archetypes=n_archs).assign(n_archetypes=n_archs))
pd.concat(df_list)
| X_pca_0 | X_pca_1 | X_pca_2 | archetype | iter | reference | mean_variance | variance_per_archetype | n_archetypes | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | -3.925065 | 0.026056 | 0.001575 | 0 | 1 | False | 0.025320 | 0.011701 | 2 |
| 1 | 6.188375 | -0.010093 | -0.157334 | 1 | 1 | False | 0.025320 | 0.038940 | 2 |
| 0 | -3.798906 | -0.199055 | 0.056105 | 0 | 2 | False | 0.025320 | 0.011701 | 2 |
| 1 | 5.991440 | 0.188029 | -0.090245 | 1 | 2 | False | 0.025320 | 0.038940 | 2 |
| 0 | -3.954038 | -0.007059 | 0.024813 | 0 | 3 | False | 0.025320 | 0.011701 | 2 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 3 | -2.455970 | -3.737336 | -2.720628 | 3 | 0 | True | 2.255955 | 3.864230 | 8 |
| 4 | -5.153388 | 3.453945 | -1.091033 | 4 | 0 | True | 2.255955 | 0.409466 | 8 |
| 5 | 3.026827 | 4.447707 | -4.195241 | 5 | 0 | True | 2.255955 | 1.723700 | 8 |
| 6 | 4.644743 | -2.341004 | 3.074518 | 6 | 0 | True | 2.255955 | 6.407274 | 8 |
| 7 | 3.629817 | -4.411222 | -4.458947 | 7 | 0 | True | 2.255955 | 0.988743 | 8 |
1785 rows × 9 columns
Archetypal Analysis#
To compute the archetypes, we can simply use the compute_archetypes function. There are many optimization parameters which we describe in more detail in the vignette about Archetypal Analysis. We only briefly demonstrate how results from archetypal analysis can be incorporated when varying parameters, using the delta parameter as an example.
To get a feeling for the archetypes we might plot the polytope on first two dimensions. However, note that we actually fitted the polytope in a higher dimensional space.
# pt.compute_archetypes(adata, n_archetypes=4, archetypes_only=False)
pt.plot_archetypes_2D(adata=adata, show_contours=True, result_filters={"n_archetypes": 4, "delta": 0.0})
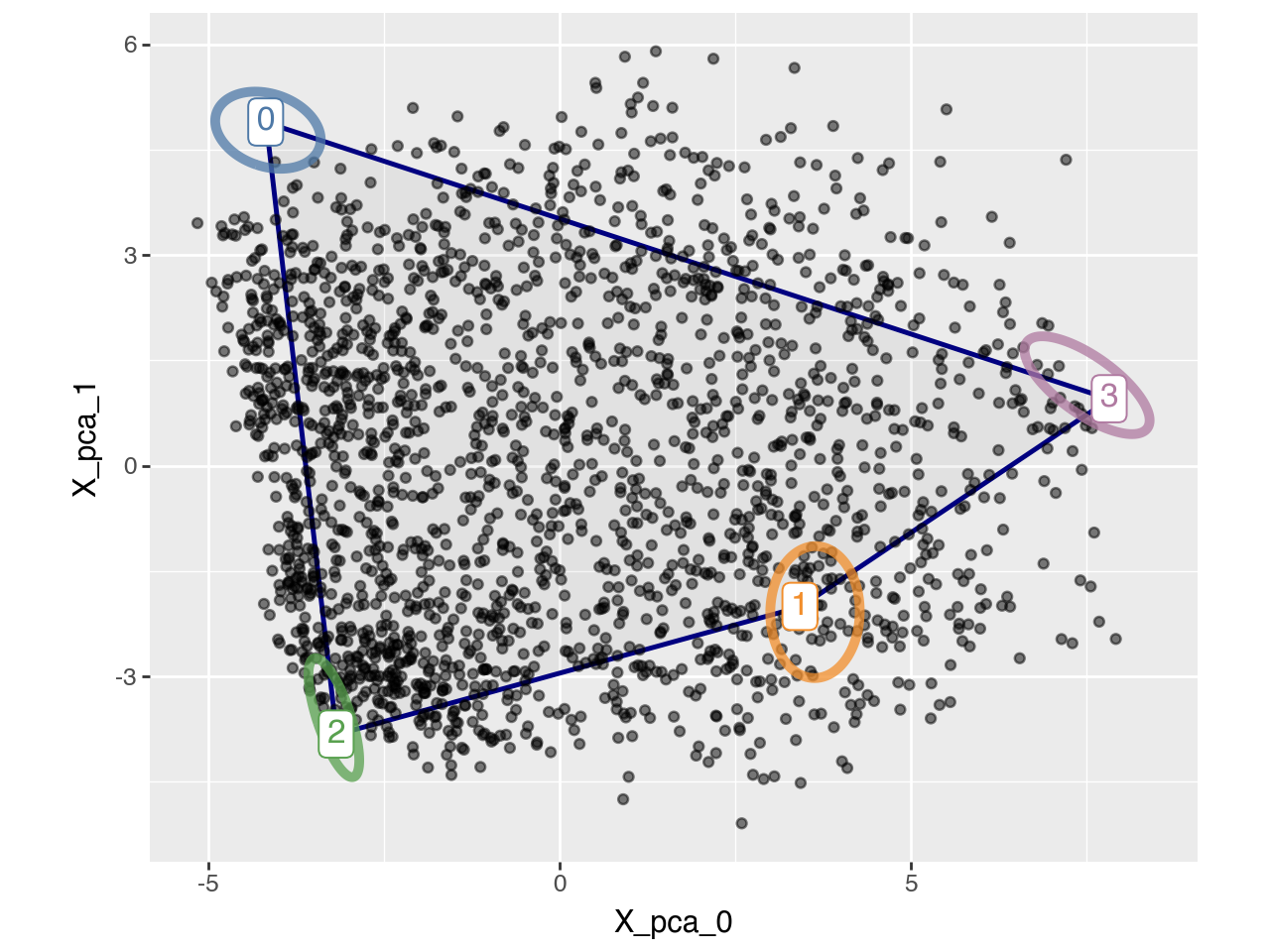
When only a few cells are sampled near the boundaries of the true phenotypic space, the convex hull of the observed data may underestimate the full range of variation. Such limited sampling can occur, for example, when the sample size is small or when the population is dominated by generalist cells rather than task-specialized ones. In these cases, relaxing the convexity constraint can help recover the true archetypes. This is achieved by introducing a relaxation parameter delta (see Equation 2 in the preprint), where larger delta values allow archetypes to lie farther from the data cloud (see Supplementary Figure S1 in the preprint). This approach has been used in [KSH+15] and subsequent works. The corresponding plot for delta=0.5 is shown below.
Whenever multiple archetypal analysis results are stored in adata.uns["AA_results"], use the result_filters dictionary to uniquely identify which result (e.g., combination of n_archetypes and delta) to visualize or analyze.
# pt.compute_archetypes(adata, n_archetypes=4, delta=0.5, archetypes_only=False)
pt.compute_bootstrap_variance(adata=adata, n_bootstrap=50, n_archetypes_list=[4], delta=0.5)
pt.plot_archetypes_2D(adata=adata, show_contours=True, result_filters={"n_archetypes": 4, "delta": 0.5})

For simplicity in this quickstart example, we will continue using delta = 0.0.
For a given \(k\), we can also visually assess bootstrap stability using pt.plot_bootstrap_2D(adata) for the first two principal components and pt.plot_bootstrap_3D(adata) for the first three.
Since Plotly does not render properly when exporting Jupyter notebooks, we will use only the 2D scatter plot here.
pt.plot_bootstrap_2D(adata, result_filters={"n_archetypes": 4, "delta": 0.0})

We can assess statistical significance by permuting the principal component (PC) coordinates and evaluating whether the polytope fit is worse on the permuted data than on the original data. To quantify goodness of fit, we can use the residual sum of squares (RSS), which returns a p-value of 0.0 in this example. Because this test used 100 permutations, this should be interpreted as an empirical lower bound (i.e., p < 0.01; equivalently < 1/101 with a +1 correction), rather than a literal zero probability. Alternatively, we can use the ratio between the volume of the convex hull of the archetypes and that of the data, and assess whether this ratio is closer to 1 for the original data than for the permuted data. Here, the resulting p-value is 0.13, meaning 13 out of 100 permutations were at least as extreme as the observed value.
significance = pt.t_ratio_significance(adata, result_filters={"n_archetypes": 4, "delta": 0.0}, n_iter=100)
significance
{'t_ratio_p_value': np.float64(0.13), 'rss_p_value': np.float64(0.0)}
Below, we will focus on the archetype that is enriched in detoxification functions.
arch_idx = 0
Characterizing Archetypes#
To better understand the archetypes, we aim to obtain the characteristic gene expression profile for each one.
We begin by computing the archetypal weights using the compute_archetype_weights() function, which assigns each cell a weight for each archetype based on its proximity to the archetype in the reduced space.
Next, we generate a the characteristic expression profile for each archetype using weighted averaging. This is achieved with the compute_archetype_expression() function, which aggregates expression data across cells according to their archetypal contributions.
To better illustrate this, we visualize the weights for archetype 0 using the plot_2D_adata() function, highlighting the extent to which each cell is associated with it.
pt.compute_archetype_weights(adata=adata, mode="automatic", result_filters={"n_archetypes": 4, "delta": 0.0})
archetype_expression = pt.compute_archetype_expression(
adata=adata, layer="z_scaled", result_filters={"n_archetypes": 4, "delta": 0.0}
)
adata.obs[f"weights_archetype_{arch_idx}"] = pt.get_aa_cell_weights(adata, n_archetypes=4, delta=0.0)[:, arch_idx]
pt.plot_archetypes_2D(
adata=adata, color=f"weights_archetype_{arch_idx}", result_filters={"n_archetypes": 4, "delta": 0.0}
)
Applied length scale is 3.25.
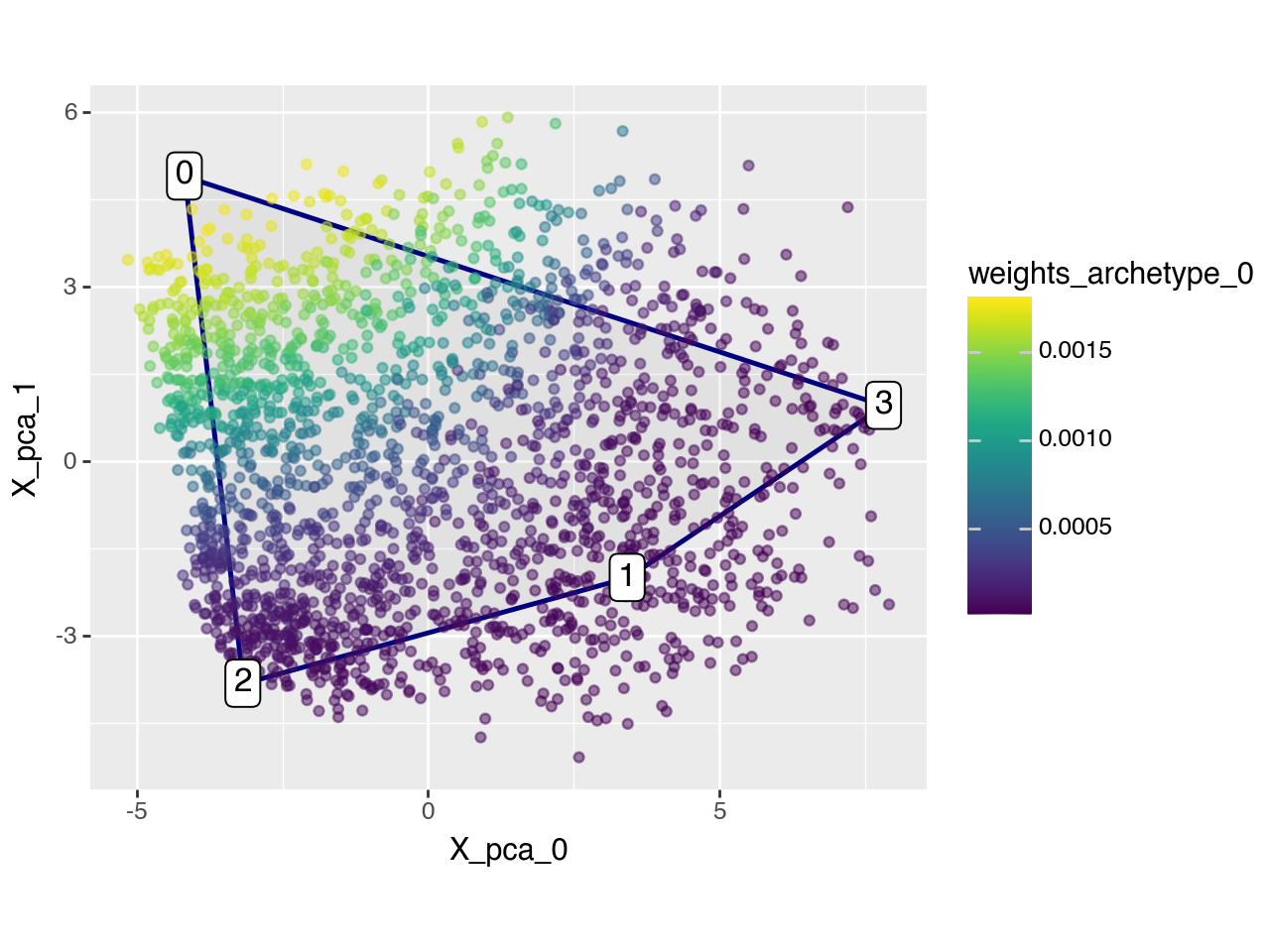
Now we can use the characteristic gene expression profile to see which genes are relatively important for each archetype. As promised above, archetype 0 is characterized by high expression of Cytochrome P450 enzymes which are important for detoxification.
archetype_expression.T.sort_values(arch_idx, ascending=False).head(10)
| 0 | 1 | 2 | 3 | |
|---|---|---|---|---|
| Cyp2e1 | 0.818915 | -0.077033 | -0.487465 | -0.128230 |
| Cyp2c29 | 0.686523 | 0.045728 | -0.443479 | -0.160054 |
| Oat | 0.678535 | -0.131683 | -0.319485 | -0.217036 |
| Rgn | 0.655820 | -0.238033 | -0.283950 | -0.134001 |
| Cyp2c50 | 0.641642 | -0.043379 | -0.376751 | -0.132530 |
| Cyp3a11 | 0.616203 | -0.070986 | -0.278696 | -0.281099 |
| Acaa1b | 0.580889 | -0.078789 | -0.319326 | -0.122232 |
| Csad | 0.576339 | -0.301767 | -0.211995 | -0.092542 |
| Cyp1a2 | 0.570929 | -0.094458 | -0.311207 | -0.105826 |
| Cyp4a10 | 0.570399 | -0.113868 | -0.240166 | -0.243191 |
We can also use the characteristic gene expression profiles of each archetype to perform enrichment analysis with decoupler. Here, we focus on Reactome pathways because we are interested in metabolic processes.
database = "reactome_pathways"
min_genes_per_pathway = 5
max_genes_per_pathway = 80
# dc.op.resource("SIGNOR", organism="mouse")
msigdb_raw = dc.op.resource("MSigDB")
msigdb = msigdb_raw[msigdb_raw["collection"] == database]
selection_vec = (
~np.array(["RESPONSE_TO" in s for s in msigdb["geneset"]])
& ~np.array(["GENE_EXPRESSION" in s for s in msigdb["geneset"]])
& ~np.array(["SARS_COV" in s for s in msigdb["geneset"]])
& ~np.array(["STIMULATED_TRANSCRIPTION" in s for s in msigdb["geneset"]])
)
msigdb = msigdb.loc[selection_vec, :].copy()
msigdb = msigdb[~msigdb.duplicated(["geneset", "genesymbol"])].copy()
genesets_within_min = (
(msigdb.value_counts("geneset") >= min_genes_per_pathway).reset_index().query("count")["geneset"].to_list()
)
genesets_within_max = (
(msigdb.value_counts("geneset") <= max_genes_per_pathway).reset_index().query("count")["geneset"].to_list()
)
genesets_to_keep = list(set(genesets_within_min) & set(genesets_within_max))
msigdb = msigdb.loc[msigdb["geneset"].isin(genesets_to_keep), :].copy() # removing small gene sets
msigdb = msigdb.rename(columns={"geneset": "source", "genesymbol": "target"}) # required since decoupler >= 2.0.0
msigdb_mouse = dc.op.translate(msigdb, target_organism="mouse", columns=["target"]) # requires decoupler >= 2.0.0
msigdb_mouse = msigdb_mouse.drop_duplicates()
acts_ulm_est, acts_ulm_est_p = dc.mt.ulm(data=archetype_expression, net=msigdb_mouse, verbose=False)
acts_ulm_est.iloc[:4, :4]
| REACTOME_2_LTR_CIRCLE_FORMATION | REACTOME_ABACAVIR_ADME | REACTOME_ABC_TRANSPORTERS_IN_LIPID_HOMEOSTASIS | REACTOME_ABERRANT_REGULATION_OF_MITOTIC_EXIT_IN_CANCER_DUE_TO_RB1_DEFECTS | |
|---|---|---|---|---|
| 0 | 0.250351 | 0.536369 | -1.368654 | -0.038143 |
| 1 | 0.790421 | 0.030883 | 2.696741 | 0.205283 |
| 2 | -0.654845 | 0.238219 | -1.966944 | -0.129916 |
| 3 | -0.332061 | -0.987201 | 1.611838 | -0.018259 |
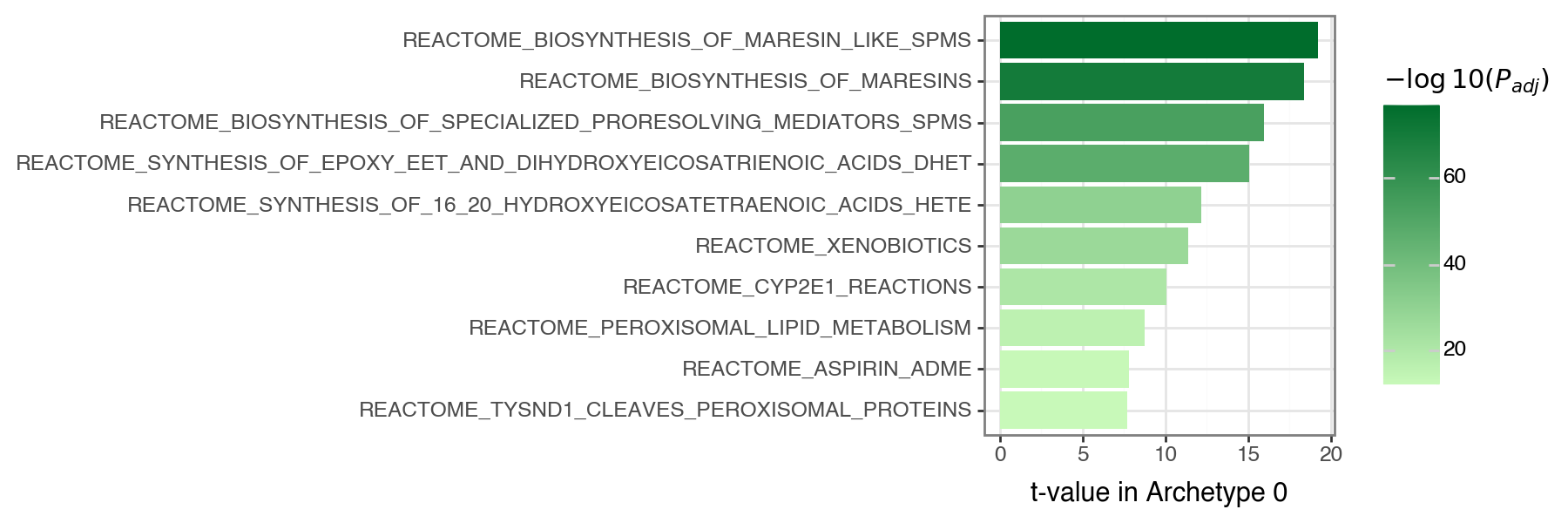
We can then extract the top \(x\) (here \(x=10\)) processes per archetype using
extract_enriched_processes(). Focusing on Archetype 0, this reveals Reactome pathways related to xenobiotics (including pathways related to oxidation and functionalization).
top_processes = pt.extract_enriched_processes(
est=acts_ulm_est, pval=acts_ulm_est_p, order="desc", n=10, p_threshold=0.05
)
top_processes[0]
| Process | 0 | 1 | 2 | 3 | specificity | act_0 | pval_0 | act_1 | pval_1 | act_2 | pval_2 | act_3 | pval_3 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | REACTOME_BIOSYNTHESIS_OF_MARESIN_LIKE_SPMS | 19.231574 | -0.073914 | -17.587075 | -3.201036 | 19.305488 | 19.231574 | 9.142448e-78 | -0.073914 | 0.997036 | -17.587075 | 4.289325e-65 | -3.201036 | 0.021992 |
| 1 | REACTOME_BIOSYNTHESIS_OF_MARESINS | 18.358449 | -0.277202 | -16.459154 | -3.236373 | 18.635651 | 18.358449 | 3.287557e-71 | -0.277202 | 0.996134 | -16.459154 | 2.617283e-57 | -3.236373 | 0.020673 |
| 2 | REACTOME_BIOSYNTHESIS_OF_SPECIALIZED_PRORESOLV... | 15.946956 | -0.924024 | -13.981915 | -2.451121 | 16.870980 | 15.946956 | 5.615380e-54 | -0.924024 | 0.866639 | -13.981915 | 1.739503e-41 | -2.451121 | 0.122326 |
| 3 | REACTOME_SYNTHESIS_OF_EPOXY_EET_AND_DIHYDROXYE... | 15.050412 | -0.072617 | -13.457141 | -2.901618 | 15.123029 | 15.050412 | 3.269266e-48 | -0.072617 | 0.997036 | -13.457141 | 1.546648e-38 | -2.901618 | 0.050339 |
| 4 | REACTOME_SYNTHESIS_OF_16_20_HYDROXYEICOSATETRA... | 12.154628 | 0.060633 | -10.380652 | -3.082588 | 12.093995 | 12.154628 | 1.735538e-31 | 0.060633 | 0.997036 | -10.380652 | 3.987993e-23 | -3.082588 | 0.031322 |
| 5 | REACTOME_XENOBIOTICS | 11.373397 | 2.173401 | -11.793347 | -2.579222 | 9.199996 | 11.373397 | 1.115591e-27 | 2.173401 | 0.303168 | -11.793347 | 1.256532e-29 | -2.579222 | 0.097644 |
| 6 | REACTOME_CYP2E1_REACTIONS | 10.048128 | 2.525538 | -10.734142 | -2.557661 | 7.522590 | 10.048128 | 1.325874e-21 | 2.525538 | 0.160444 | -10.734142 | 1.092435e-24 | -2.557661 | 0.100943 |
| 7 | REACTOME_PEROXISOMAL_LIPID_METABOLISM | 8.744138 | -2.767583 | -5.231965 | -1.933989 | 10.678127 | 8.744138 | 1.862506e-16 | -2.767583 | 0.092332 | -5.231965 | 3.403079e-06 | -1.933989 | 0.289015 |
| 8 | REACTOME_ASPIRIN_ADME | 7.787722 | 1.642603 | -7.991401 | -1.944230 | 6.145119 | 7.787722 | 3.972944e-13 | 1.642603 | 0.595242 | -7.991401 | 9.677742e-14 | -1.944230 | 0.287879 |
| 9 | REACTOME_TYSND1_CLEAVES_PEROXISOMAL_PROTEINS | 7.687112 | -1.734315 | -5.439396 | -1.463539 | 9.150652 | 7.687112 | 7.745794e-13 | -1.734315 | 0.547397 | -5.439396 | 1.143445e-06 | -1.463539 | 0.509333 |
top_processes[arch_idx]
| Process | 0 | 1 | 2 | 3 | specificity | act_0 | pval_0 | act_1 | pval_1 | act_2 | pval_2 | act_3 | pval_3 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | REACTOME_BIOSYNTHESIS_OF_MARESIN_LIKE_SPMS | 19.231574 | -0.073914 | -17.587075 | -3.201036 | 19.305488 | 19.231574 | 9.142448e-78 | -0.073914 | 0.997036 | -17.587075 | 4.289325e-65 | -3.201036 | 0.021992 |
| 1 | REACTOME_BIOSYNTHESIS_OF_MARESINS | 18.358449 | -0.277202 | -16.459154 | -3.236373 | 18.635651 | 18.358449 | 3.287557e-71 | -0.277202 | 0.996134 | -16.459154 | 2.617283e-57 | -3.236373 | 0.020673 |
| 2 | REACTOME_BIOSYNTHESIS_OF_SPECIALIZED_PRORESOLV... | 15.946956 | -0.924024 | -13.981915 | -2.451121 | 16.870980 | 15.946956 | 5.615380e-54 | -0.924024 | 0.866639 | -13.981915 | 1.739503e-41 | -2.451121 | 0.122326 |
| 3 | REACTOME_SYNTHESIS_OF_EPOXY_EET_AND_DIHYDROXYE... | 15.050412 | -0.072617 | -13.457141 | -2.901618 | 15.123029 | 15.050412 | 3.269266e-48 | -0.072617 | 0.997036 | -13.457141 | 1.546648e-38 | -2.901618 | 0.050339 |
| 4 | REACTOME_SYNTHESIS_OF_16_20_HYDROXYEICOSATETRA... | 12.154628 | 0.060633 | -10.380652 | -3.082588 | 12.093995 | 12.154628 | 1.735538e-31 | 0.060633 | 0.997036 | -10.380652 | 3.987993e-23 | -3.082588 | 0.031322 |
| 5 | REACTOME_XENOBIOTICS | 11.373397 | 2.173401 | -11.793347 | -2.579222 | 9.199996 | 11.373397 | 1.115591e-27 | 2.173401 | 0.303168 | -11.793347 | 1.256532e-29 | -2.579222 | 0.097644 |
| 6 | REACTOME_CYP2E1_REACTIONS | 10.048128 | 2.525538 | -10.734142 | -2.557661 | 7.522590 | 10.048128 | 1.325874e-21 | 2.525538 | 0.160444 | -10.734142 | 1.092435e-24 | -2.557661 | 0.100943 |
| 7 | REACTOME_PEROXISOMAL_LIPID_METABOLISM | 8.744138 | -2.767583 | -5.231965 | -1.933989 | 10.678127 | 8.744138 | 1.862506e-16 | -2.767583 | 0.092332 | -5.231965 | 3.403079e-06 | -1.933989 | 0.289015 |
| 8 | REACTOME_ASPIRIN_ADME | 7.787722 | 1.642603 | -7.991401 | -1.944230 | 6.145119 | 7.787722 | 3.972944e-13 | 1.642603 | 0.595242 | -7.991401 | 9.677742e-14 | -1.944230 | 0.287879 |
| 9 | REACTOME_TYSND1_CLEAVES_PEROXISOMAL_PROTEINS | 7.687112 | -1.734315 | -5.439396 | -1.463539 | 9.150652 | 7.687112 | 7.745794e-13 | -1.734315 | 0.547397 | -5.439396 | 1.143445e-06 | -1.463539 | 0.509333 |
top_processes[arch_idx]["Process"] = pd.Categorical(
top_processes[arch_idx]["Process"],
categories=top_processes[arch_idx].sort_values(f"{arch_idx}", ascending=True)["Process"].to_list(),
)
top_processes[arch_idx][f"neg_log10_pval_{arch_idx}"] = -np.log10(top_processes[arch_idx][f"pval_{arch_idx}"])
p = (
pn.ggplot(top_processes[arch_idx])
+ pn.geom_col(pn.aes(x="Process", y=f"act_{arch_idx}", fill=f"neg_log10_pval_{arch_idx}"))
+ pn.coord_flip()
+ pn.theme_bw()
+ pn.theme(figure_size=(9, 3))
+ pn.labs(x="", y=f"t-value in Archetype {arch_idx}", fill=r"$-\log10(P_{adj})$")
+ pn.scale_fill_gradient(low="#c8f9b9", high="#006d2c")
)
p.show()
Then, we can decoupler’s leading edge function to check for example which genes have a high weight for our archetype of interest, and are in the gene set for “REACTOME_XENOBIOTICS”.
df = archetype_expression.T.copy()
df.columns = df.columns.astype(str)
_, le = dc.pl.leading_edge(df=df, net=msigdb_mouse, stat=str(arch_idx), name="REACTOME_XENOBIOTICS")
print(le)
['Cyp2e1' 'Cyp2c29' 'Cyp2c50' 'Cyp1a2' 'Cyp2c37' 'Cyp2a5' 'Cyp2c38' 'Ahr']
Another interesting thing we can do is to consider how gene expressions changes as a function of the distance to some archetype. To this end we can use the decoupler functions dc.tl.rankby_order, dc.pp.bin_order, and dc.pl.order.
Z = pt.get_aa_result(adata, n_archetypes=4, delta=0)["Z"]
distances = cdist(adata.obsm[adata.uns["AA_config"]["obsm_key"]][:, adata.uns["AA_config"]["n_dimensions"]], Z)
for idx in range(Z.shape[1]):
adata.obs[f"dist_to_arch_{idx}"] = distances[:, idx]
genes_ordered = dc.tl.rankby_order(
adata=adata,
order=f"dist_to_arch_{arch_idx}",
stat="spearmanr",
)
top_genes = genes_ordered.sort_values("stat").head(8)["name"].to_list()
bottom_genes = genes_ordered.sort_values("stat").tail(8)["name"].to_list()
top_genes_ordered = dc.pp.bin_order(
adata=adata,
order=f"dist_to_arch_{arch_idx}",
names=top_genes,
nbins=50,
)
bottom_genes_ordered = dc.pp.bin_order(
adata=adata,
order=f"dist_to_arch_{arch_idx}",
names=bottom_genes,
nbins=50,
)
dc.pl.order(
df=top_genes_ordered,
mode="line",
figsize=(6, 3),
)
dc.pl.order(
df=bottom_genes_ordered,
mode="line",
figsize=(6, 3),
)
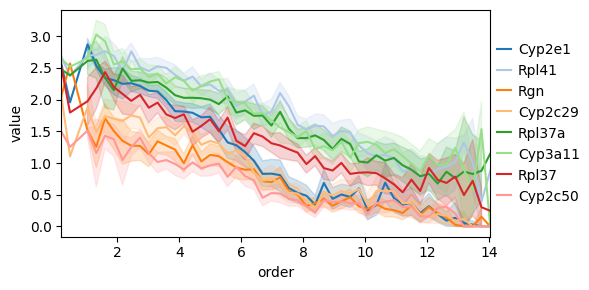
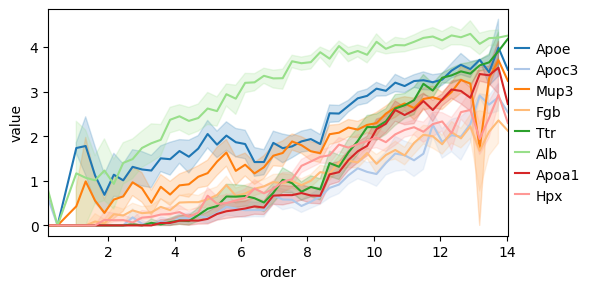
Alternatively, we can use the quantile-based, distance-ranking gene-enrichment algorithm used in previous ParTI work [AKKT+19, HSH+15, KSH+15] (see Supplementary Methods Section 4.4), which tests for genes whose expression is highest in the quantile bin closest to an archetype relative to a background (by default all remaining cells, with optional alternative contrasts such as closest versus furthest bins). This algorithm is implemented in pt.compute_quantile_based_gene_enrichment.
However, note that this approach requires per-gene and per-archetype hypothesis testing across large numbers of cells and therefore scales less well than kernel-aggregated profiling.
gene_enrichment = pt.compute_quantile_based_gene_enrichment(adata, result_filters={"n_archetypes": 4, "delta": 0.0})
gene_enrichment.query("enriched").query(f"arch_idx=={arch_idx}").sort_values("pval_adj").head(6)
| arch_idx | gene | stat | pval | mean_diff | median_diff | mean_bin0 | mean_rest | median_bin0 | median_rest | max_in_bin0 | pos_effect | pval_adj | signif | enriched | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3779 | 0 | Oat | 306028.5 | 7.122084e-85 | 0.998351 | 1.177536 | 1.253019 | 0.254668 | 1.177536 | 0.000000 | True | True | 5.949789e-81 | True | True |
| 3801 | 0 | Cyp2e1 | 315818.0 | 3.467040e-73 | 1.352518 | 2.014842 | 2.181629 | 0.829111 | 2.221061 | 0.206219 | True | True | 9.654550e-70 | True | True |
| 8235 | 0 | Cyp2c50 | 289663.0 | 1.178018e-48 | 0.678192 | 0.807465 | 1.207931 | 0.529739 | 1.236465 | 0.429001 | True | True | 1.968233e-45 | True | True |
| 4263 | 0 | Rpl41 | 293127.0 | 1.654406e-48 | 0.871232 | 0.713324 | 2.552605 | 1.681374 | 2.618408 | 1.905084 | True | True | 2.303484e-45 | True | True |
| 8227 | 0 | Cyp2c29 | 290362.5 | 2.534837e-48 | 0.896444 | 1.324256 | 1.629166 | 0.732722 | 1.712150 | 0.387894 | True | True | 3.025147e-45 | True | True |
| 2009 | 0 | Cyp4a10 | 272953.0 | 4.374906e-48 | 0.541017 | 0.781933 | 0.764036 | 0.223019 | 0.781933 | 0.000000 | True | True | 4.568496e-45 | True | True |
Spatial Enrichment#
As in [AKKT+19], we want to quantify where the archetypes locate in the tissue.
To that end, we download a single-cell spatial dataset from the liver from https://info.vizgen.com/mouse-liver-data (Vizgen MERFISH Mouse Liver Map January 2022). In particular, we only need to download from Liver1Slice1 the files "cell_by_gene.csv", "cell_metadata.csv" and place them in the folder Liver1Slice1 in data.
Then, to quantify which archetype any given cell is close to, we compute the enrichment of the archetype gene expression signatures in each cell’s gene expression profile.
# only run if the data has actually been downloaded
if (Path(".") / "data" / "Liver1Slice1" / "Liver1Slice1_cell_by_gene.csv").exists() and (
Path(".") / "data" / "Liver1Slice1" / "Liver1Slice1_cell_metadata.csv"
).exists():
import squidpy as sq
vizgen_adata = sq.read.vizgen(
"./data/Liver1Slice1",
counts_file="Liver1Slice1_cell_by_gene.csv",
meta_file="Liver1Slice1_cell_metadata.csv",
)
vizgen_adata = vizgen_adata[:, vizgen_adata.var.index.isin(archetype_expression.columns.to_list())].copy()
vizgen_adata = vizgen_adata[vizgen_adata.X.sum(axis=1).A1 > 200, :].copy()
sc.pp.normalize_total(vizgen_adata)
sc.pp.log1p(vizgen_adata)
# create weights based on archetype expression profile
archetype_expression_weights = (
archetype_expression[vizgen_adata.var.index]
.copy()
.reset_index(names="archetype")
.melt(id_vars="archetype", value_name="weight", var_name="gene")
)
archetype_expression_weights = archetype_expression_weights.rename(
columns={"archetype": "source", "gene": "target"}
)
# compute the enrichment of the archetype gex profiles in the gex profiles of each cell
dc.mt.ulm(data=vizgen_adata, net=archetype_expression_weights, verbose=False)
# plotting one part
plotting_df = vizgen_adata.obsm["score_ulm"].copy()
plotting_df.columns = ["archetype_" + idx for idx in plotting_df.columns]
plotting_df = plotting_df - plotting_df.mean()
plotting_df = plotting_df / plotting_df.std()
plotting_df["x"] = vizgen_adata.obsm["spatial"][:, 0]
plotting_df["y"] = vizgen_adata.obsm["spatial"][:, 1]
width = 3_000
x_low = 5_000
y_low = 6_000
x_high = x_low + width
y_high = y_low + width
plotting_df = plotting_df.loc[
(plotting_df["x"] > x_low)
& (plotting_df["x"] < x_high)
& (plotting_df["y"] > y_low)
& (plotting_df["y"] < y_high)
]
plotting_df = plotting_df.melt(id_vars=["x", "y"], value_name="score", var_name="archetype")
p = (
pn.ggplot(plotting_df)
+ pn.geom_point(pn.aes(x="x", y="y", color="score"), size=0.1, alpha=0.5)
+ pn.scale_color_gradient2(low="blue", mid="white", high="red", midpoint=0)
+ pn.facet_wrap(facets="archetype", nrow=2, ncol=2)
+ pn.coord_equal()
+ pn.theme(figure_size=(8, 8))
+ pn.labs(x="x", y="y", color="Activity")
)
p.show()
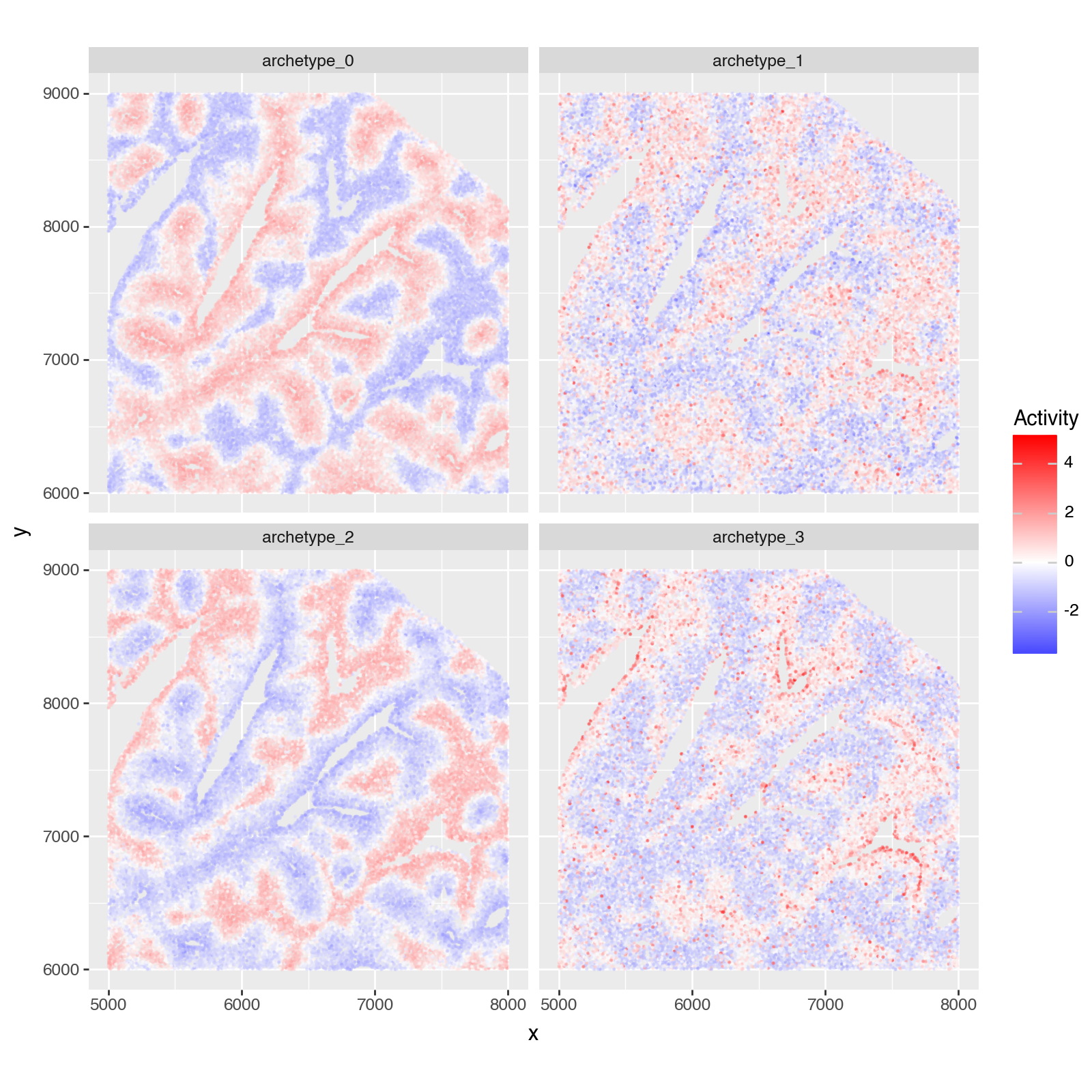
Saving and Loading Results#
See more details in the Accessing And Saving Cached Results vignette vignette. In brief: ParTIpy caches AA outputs with immutable ArchetypeConfig keys (a frozen Pydantic model), those objects keep the cache hashable inside adata.uns. HDF5 only accepts string keys, so pt.write_h5ad turns each config into a JSON string before writing, and pt.read_h5ad restores the original ArchetypeConfig objects so cached metrics still work after reloading.
pt.write_h5ad(adata, "adata.h5ad")
adata = pt.read_h5ad("adata.h5ad")
adata
AnnData object with n_obs × n_vars = 1999 × 8354
obs: 'cell_type', 'zone', 'run_id', 'time_point', 'UMAP_X', 'UMAP_Y', 'weights_archetype_0', 'dist_to_arch_0', 'dist_to_arch_1', 'dist_to_arch_2'
var: 'highly_variable', 'means', 'dispersions', 'dispersions_norm'
uns: 'AA_bootstrap', 'AA_cell_weights', 'AA_config', 'AA_pca', 'AA_results', 'AA_selection_metrics', 'AA_t_ratio', 'hvg', 'log1p', 'pca'
obsm: 'X_pca'
varm: 'PCs'
layers: 'z_scaled'
Session Info#
import session_info2
print(session_info2.session_info(dependencies=True))
pandas 2.2.3
scanpy 1.11.2
plotnine 0.14.5
decoupler 2.0.6
numpy 2.2.6
scipy 1.15.2
anndata 0.11.4
squidpy 1.6.5
---- ----
jaraco.text 3.12.1
geopandas 1.0.1
annotated-types 0.7.0
param 2.2.0
adjustText 1.3.0
zarr 2.18.7
importlib_metadata 8.7.0
asttokens 3.0.0
tifffile 2025.5.26
zstandard 0.25.0
imageio 2.37.0
cycler 0.12.1
pydantic_core 2.41.1
crc32c 2.7.1
validators 0.35.0
multiscale_spatial_image 2.0.2
debugpy 1.8.14
setuptools 80.8.0
pyct 0.5.0
pure_eval 0.2.3
ipywidgets 8.1.7
numcodecs 0.15.1
multipledispatch 1.0.0 (0.6.0)
coverage 7.10.7
stack-data 0.6.3
six 1.17.0
networkx 3.5
cloudpickle 3.1.1
matplotlib-scalebar 0.9.0
pyproj 3.7.1
marsilea 0.5.3
dcor 0.6
python-dateutil 2.9.0.post0
parso 0.8.4
MarkupSafe 3.0.3
PyYAML 6.0.2
jaraco.classes 3.4.0
fsspec 2025.5.1
wcwidth 0.2.13
statsmodels 0.14.4
zipp 3.22.0
more-itertools 10.7.0
attrs 25.3.0
wrapt 1.17.2
narwhals 1.41.0
ome-zarr 0.11.1
asciitree 0.3.3
scikit-learn 1.6.1
idna 3.10
packaging 25.0
colorama 0.4.6
mizani 0.13.5
Pygments 2.19.1
spatial_image 1.2.1
legendkit 0.3.5
ipython 9.2.0
xarray-dataclasses 1.9.1
session-info2 0.1.2
appnope 0.1.4
jedi 0.19.2
requests 2.32.3
dask-image 2024.5.3
pyparsing 3.2.3
datashader 0.18.1
threadpoolctl 3.6.0
charset-normalizer 3.4.2
docrep 0.3.2
platformdirs 4.3.8
seaborn 0.13.2
pytz 2025.2
certifi 2025.4.26 (2025.04.26)
numba 0.61.2
jaraco.context 6.0.1
plotly 6.1.2
prompt_toolkit 3.0.51
spatialdata 0.4.0
scikit-image 0.25.2
pyzmq 26.4.0
lazy_loader 0.4
matplotlib 3.10.3
xarray 2024.11.0
jaraco.collections 5.1.0
igraph 0.11.8
torch 2.10.0
xarray-spatial 0.4.0
natsort 8.4.0
xarray-schema 0.0.3
pillow 11.2.1
llvmlite 0.44.0
jupyter_core 5.8.1
traitlets 5.14.3
urllib3 2.4.0
jaraco.functools 4.3.0
pydantic 2.12.0
texttable 1.7.0
comm 0.2.2
Jinja2 3.1.6
legacy-api-wrap 1.4.1
Deprecated 1.2.18
typing_extensions 4.15.0
joblib 1.5.1
patsy 1.0.1
tornado 6.5.1
rich 14.0.0
toolz 1.0.0
typing-inspection 0.4.2
pyarrow 20.0.0
executing 2.2.0
xgboost 3.0.2
dask 2024.11.2
ipykernel 6.29.5
kiwisolver 1.4.8
matplotlib-inline 0.1.7
jupyter_client 8.6.3
tqdm 4.67.1
decorator 5.2.1
h5py 3.13.0
shapely 2.1.1
psutil 7.0.0
---- ----
Python 3.11.0 | packaged by conda-forge | (main, Jan 14 2023, 12:26:40) [Clang 14.0.6 ]
OS macOS-15.7.1-arm64-arm-64bit
Updated 2026-02-17 09:39